KEY TENETS OF THE U.S. BIOSIMILARS REGULATORY PATHWAY
In the U.S., the regulatory pathway for all medicines, including biosimilars, is rigorous. At the core of the biosimilars regulatory pathway is the mandate that biosimilar sponsors must demonstrate that the biosimilar is highly similar to the reference product notwithstanding minor differences that may exist in clinically inactive components, and that there are no clinically meaningful differences between the biosimilar and the reference product in terms of safety, purity, and potency.
The FDA regulatory pathway accounts for safety and efficacy for biosimilars in an analogous manner to the European Union, where biosimilars have been approved for use since 2006. As of mid-2017, Europe had approved 38 different biosimilar products, which are now marketed under 39 different brand names. As a result, in Europe, clinicians and patients have benefited from the safe use of millions of doses of biosimilar medicines. As of July 2017, there have been more than 700 million patient days of experience with biosimilars in Europe (Regulations.gov, Biosimilar Medicines Group, Medicine for Europe sector group, June 26, 2017). In the more than ten years since biosimilars were first marketed in Europe, European regulators have concluded that there have been no additional unexpected safety concerns about biosimilars that were not already known from the reference product.
The Biologics Price Competition and Innovation Act of 2009 (BPCI Act) was signed into law in 2010 as a part of the Patient Protection and Affordable Care Act (ACA). The ACA was enacted to increase the quality and affordability of health care while reducing costs for individuals and the government. As part of this, an abbreviated licensure pathway was created for biological products shown to be biosimilar to or interchangeable with an FDA-licensed reference product. There is broad bipartisan support in Congress and in the health care community for the BPCIA.
Definition of Biosimilar
According to the FDA, a biosimilar is a biological product that has been proven to be highly similar to a reference product notwithstanding minor differences in clinically inactive components, and that there are no clinically meaningful differences between the biosimilar and the reference product in terms of safety, purity, and potency. Patients and their healthcare providers can expect the same clinical outcomes when using a biosimilar as would be expected by use of the reference product.
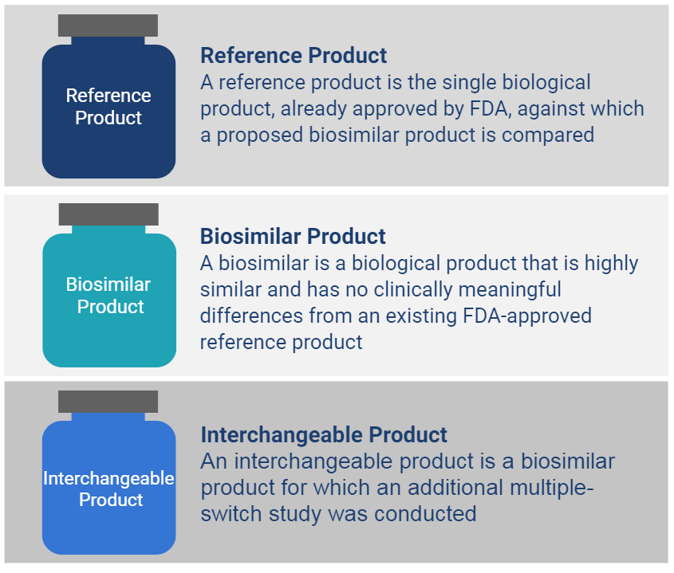
Definition of Interchangeable
In the U.S., when the FDA determines a product to be interchangeable with its reference product, it can be expected to produce the same clinical result as the reference product in any given patient. It must be biosimilar and also demonstrate through additional clinical data, that the risk in terms of safety or diminished efficacy of alternating or switching between use of the interchangeable product and its reference product is not greater than the risk of using the reference product without alternating or switching.
The designation “interchangeable” or “interchangeability,” in reference to a biological product that is shown to meet the standards described in the Public Health Service Act, means that the biological product may be substituted by a pharmacist for the reference product without first getting permission from the health care provider who prescribed the reference product, subject to state substitution laws.
There is no difference in quality between biosimilars that are interchangeable and those that are not interchangeable. The quality requirements for both are the same.
By definition, biosimilars in the U.S. must be approved by use of the FDA’s Public Health Service Act Section 351(k) application. This is different than the 351(a) Biological License Application (BLA) pathway of the Public Health Services Act that is used for originator biological drugs. Different regulatory pathways are used for chemical drugs and for small-molecule generic drugs.
A 351(k) application must include information demonstrating that:

The data demonstrating biosimilarity must be derived from:

Data demonstrating interchangeability must demonstrate that if a biosimilar product that is administered more than once to an individual the risk in terms of safety or diminished efficacy of alternating or switching between the biosimilar and its reference product is not greater than the risk of using the reference product without alternating or switching.
In the application process, the FDA may determine, at its discretion and on a product-by-product basis, that an element in the application process described above is unnecessary for a specific 351(k) application.
Even though the 351(k) application is a different regulatory pathway than that the 351(a) regulatory pathway used for the reference product, the quality requirements and expectations for biosimilars and interchangeable biologics are identical to those of the biological reference product. The quality requirements and expectations include all aspects of the manufacturing process, the features of the biological drugs and their stability.
Following submission of the complete 351(k) application, the FDA confirms that the product is biosimilar to or interchangeable with the reference product. The facility where the biosimilar is inspected by the FDA to ensure that it complies with good manufacturing practice (GMP) requirements that are identical to those applied to all biological drugs. Once the FDA review is complete and all requirements are met to the satisfaction of the FDA, the product is then licensed for its designated therapy.