BIOSIMILARS FAQS
Here are answers to some of the most common questions people have about biosimilars and how they’ll be used and regulated in the U.S. If you don’t find the information you are seeking, please contact us.
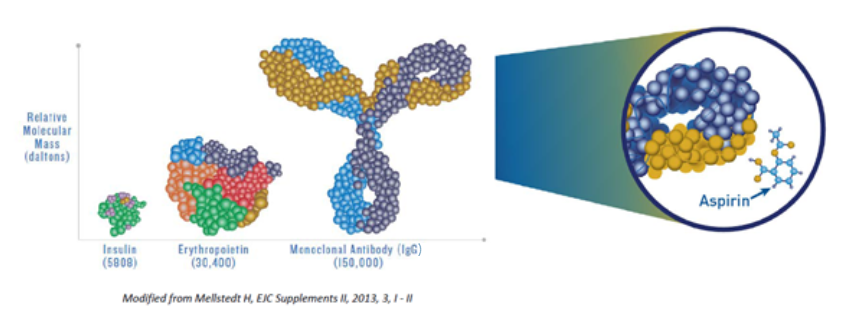
Biological medicines, also known as biological therapies or “biologics,” are medicines that are produced by living organisms, including human, animal, or microorganism cells. Biologics are very large, complex molecules or mixtures of molecules and can be proteins, sugars, and nucleic acids, as well as living entities such as cells and tissues. Today, they are commonly produced through biotechnology.
Biologics are different than most drugs used today because most drugs are made by synthetic chemical processes and are much smaller molecules than biologics.
Biologics work within a patient’s body by supplementing or interrupting natural pathways, processes, and signals in order to treat a specific disease or disorder.
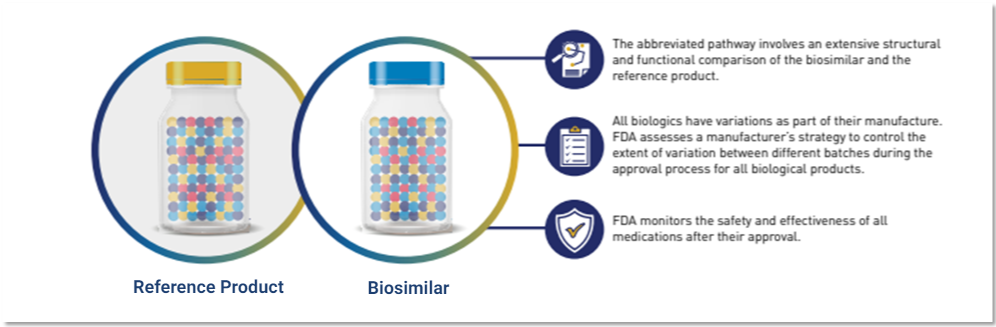
All biological medicines vary to a degree within a batch as well as from one batch to the next when the same manufacturing process is used. This is normal and expected. Rigorous manufacturing controls are put in place to ensure that all variability stays within pre-specified and pre-approved ranges to ensure that the clinical benefit and safety are unchanged over time.
(Image source: U.S. FDA)
A biosimilar medicine is a biologic that is approved after it is demonstrated to be highly similar to an FDA-approved biological medicine, known as a reference product. A biosimilar must be determined by the FDA to (1) be highly similar to the reference product notwithstanding minor differences in clinically inactive components and (2) have no clinically meaningful differences compared to the reference product in terms of safety, purity, and potency.
Biosimilars will have the same safety and effectiveness as the reference product.
Biosimilars are neither required nor expected to be “identical” to the reference biologic, which itself has some degree of inherent variability because they are produced in living cells. Nonetheless, it is important to know that biosimilars are highly similar to the reference product, and any differences that may exist will not have an impact on safety or effectiveness.
When applied to biological drugs, the term “interchangeable” or “interchangeability,” means that a biosimilar drug may be substituted at the pharmacy counter by a pharmacist for the reference product without first getting permission from the health care provider – typically a physician or nurse practitioner – who wrote the prescription. Interchangeability is different from substitution that is initiated by the health care provider that occurs when a healthcare provider writes a prescription for one drug in place of another. Physicians always have the freedom to prescribe whatever drug they believe is appropriate for their patients.
According to U.S. law, an interchangeable biologic is a distinct regulatory category of biosimilar and is not a different product. To obtain an FDA designation of “interchangeability,” the sponsor of a biosimilar typically must conduct an additional clinical study with multiple switches back and forth between the biosimilar and reference product. This clinical study must prove that for a product administered to an individual more than once, the risk in terms of safety or diminished efficacy of switching back and forth between the interchangeable biologic and its reference product is not greater than the risk of using the reference product by itself, without switching.
The interchangeable biologic molecule is exactly the same as the biosimilar that was initially approved from that sponsor, — the difference being that additional clinical data was provided in order to gain the interchangeability designation from the FDA. There is no difference in quality.
While the FDA will designate biologic interchangeability nationally, individual states regulate the practice of pharmacy, including laws describing how and when a pharmacist can substitute one drug for another. State substitution laws permitting pharmacy-level substitution of an interchangeable biologic for a reference product are alike from state-to-state, but there are minor differences. Details of individual state substitution laws can be located here.
Some of the most difficult-to-treat diseases, such as cancer, anemia, multiple sclerosis, and autoimmune disorders (e.g., rheumatoid arthritis, psoriasis, and inflammatory bowel disease) may be treated or managed with biologics and biosimilars. Biologic and biosimilar treatments are delivered in a in the form of an injectable administered by the patient, care-giver, or health care professional or as a solution to be administered intravenously. While biologics make up a small percentage of the total number of drugs on the market, they can be very expensive to the patients who rely on them and are a significant cost to the U.S. health care system.
Biosimilars can save $133 billion by 2025 if their use increases.
Since launch of the first biosimilar in the US in 2015, competition from biosimilars has lowered prices of both biosimilars and their reference biologics .
Biosimilars have lowered the cost of the reference brand biologic by 30%.

The growth of the biosimilar market will provide multiple sourcing options for health care professionals and may provide broader and earlier patient access to these important treatment options. Since launch of the first biosimilar in the US in 2015, competition from biosimilars has lowered prices of both biosimilars and their reference biologics.
This has been observed in some European markets where the number of patients being treated with a given biologic (combined originator and biosimilar) has increased since the introduction of biosimilars in Europe more than ten years ago.
Biosimilars provide more treatment options and may help to bring down prices for patients. The availability of affordable biosimilars may improve access and enable more patients to be treated with these therapies. Data have shown that in some indications for which biologics may be used, the use of biologics earlier in a treatment regimen (e.g. by lessening or eliminating step therapy) may improve individual patient outcomes when compared to patients not treated with biologics until later in the treatment cycle.
Lower medication costs due to biosimilars may lessen the financial strain for some patients, making it easier for them to remain on therapy longer.
The introduction of biosimilars on the market increases treatment options and competition. This competition may lead to high-value therapies at reduced prices. Industry experts have suggested the savings obtained with biosimilars could free up health care funding for coverage of new medicines or other societal needs.
Lower medication costs due to biosimilars may make it easier for some patients to remain on therapy longer which could help avoid more intensive healthcare resource utilization that might occur if patients do not adhere to their medication therapy.
Since launch of the first biosimilar in the US in 2015, competition from biosimilars has lowered prices of both biosimilars and their reference biologics.
Yes, patients and their healthcare providers can expect the same benefits and potential side effects when using a biosimilar as would be expected using the biological reference product. There are no clinically meaningful differences in terms of safety, purity, and potency. A biosimilar must have the same mechanism(s) of as its reference product to the extent they are known, which means it will work in the same way in the body. The FDA will only approve a biosimilar if:
-
- Convincing data is provided to demonstrate that it is highly similar to the reference product.
- It utilizes the same mechanism(s) of action for the condition or conditions of use prescribed (to the extent they are known for the reference product).
- Condition(s) of use have been previously approved for the reference product. Route of administration, dosage form, and the strength of the biological product are the same as those of the reference product.
- The facility in which the biosimilar is manufactured, processed, packed, or stored meets standards designed to assure that the biological product continues to be safe, pure, and potent.
Biosimilars are held to the very same rigorous manufacturing and quality standards as are applied to their reference products.
(Image source: U.S. FDA)
A biosimilar may be prescribed by a physician for both patients newly diagnosed with a disease, and for patients who may already have been treated with the reference product. A provider must write the specific name of the biosimilar on the prescription or order. A pharmacist must dispense the biologic specified or must obtain permission in advance from the prescribing physician to dispense a different biologic in place of the product identified on the prescription order.
If a biosimilar is also approved by the FDA as an interchangeable biologic it may be substituted by a pharmacist for the prescribed reference product without first getting permission from of the prescribing physician, subject to the appropriate state laws governing the practice of pharmacy.

No. Biosimilars are not like generic drugs because most generic drugs are chemically synthesized while biosimilars are manufactured using living systems. Generic drugs are identical copies of brand-name drugs, and sponsors must demonstrate that generic drug levels in the body are the same over time as their respective reference drugs. Generic drugs have the identical active ingredient and are the same in terms of dose form, safety, strength, administration, quality, performance characteristics, and intended use.
In contrast to generics, biosimilars are derived from living systems and are commonly more complex than generic drugs. Biosimilars are highly similar to their respective reference product, even though there may be minor differences in clinically inactive components. Manufacturers must demonstrate that there are no clinically meaningful differences between a biosimilar and reference product in terms of safety, purity, and potency.
However, as with generic drugs, patients and their healthcare providers can expect the same benefits and potential side effects when using a biosimilar as would be expected using the biological reference product.
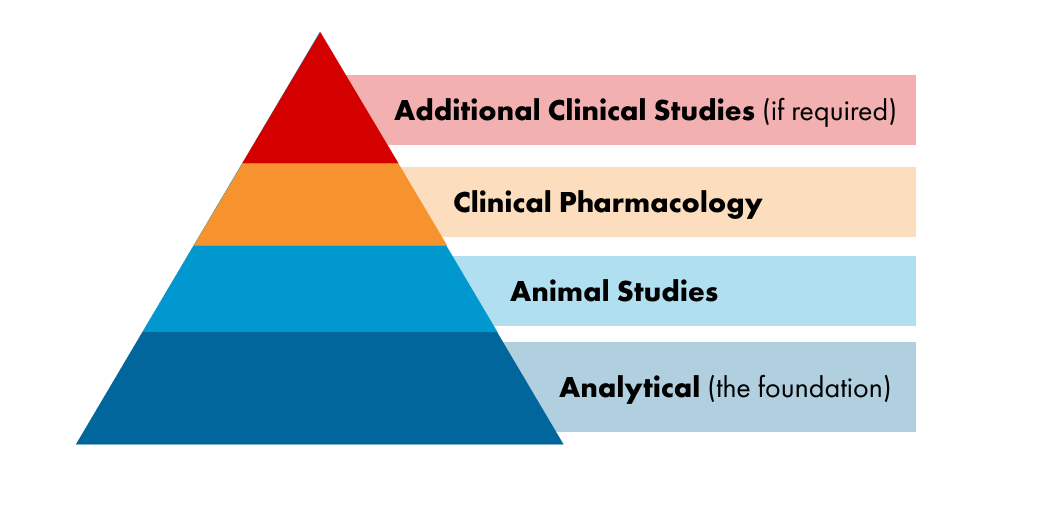
Biosimilars are approved by the FDA using a regulatory pathway that is different than that used for new chemical and biological drugs. The biosimilar regulatory pathway focuses heavily on analytical testing that consists of structural analysis and measures of biological activity that are made in a laboratory setting. These are also known as biological function assays. Some clinical studies are often required, but clinical studies primarily serve to confirm the high similarity that is seen with analytical testing. The heavy focus on structural and analytical testing is appropriate because analytics are often more sensitive and have a greater capability to detect clinically significant differences in structure and function than would be apparent in a clinical study.
Biosimilars are approved using data obtained in multiple and extensive, head-to-head comparisons of the biosimilar and reference product, which may include:

These data, taken together, form the “totality of evidence” that the FDA examines when considering approval of a biosimilar, although the FDA has the discretion to decide that certain types data may not be needed for a particular biosimilar.
Once satisfied that the totality of evidence is both substantial and sensitive enough to detect potential differences and to address residual uncertainty that may exist, the FDA could approve the biosimilar. The biosimilar regulatory pathway specifies that a biosimilar can be approved for use in one or more indications of use on the basis of the biosimilar being highly similar to the reference product, notwithstanding minor differences in clinically inactive components, and that there are no clinically meaningful differences between the biosimilar and the reference product in terms of safety, purity, and potency.
Once approved, patients and their healthcare providers can expect the same benefits and potential side effects when using a biosimilar as would be expected using the biological reference product.
Since biosimilars began to be developed almost twenty years ago, there have been significant advances in the methods used to develop biosimilars to better match the structural features of the reference product. At the same time, the methods to analyze biological drugs have advanced, enabling us to better detect and measure any differences that may exist between a biosimilar and its reference product.
These advances provide an opportunity to reconsider the requirements to establish biosimilarity by applying the best possible science. The FDA and industry have agreed to a regulatory science project which will begin in late 2022 that will evaluate opportunities to streamline biosimilar development, potentially accelerating patient access to these medicines. The Biosimilars Forum strongly supports this regulatory science project.